
Duchenne Muscular Dystrophy (DMD) is caused by mutations in the dystrophin gene located on the X-chromosome. Due to the large size of the dystrophin gene, it is highly susceptible to spontaneous mutation. In fact, thirty percent of the mutations in the dystrophin gene arise spontaneously, which means that DMD disease will always be present in the population. DMD is the most prevalent lethal genetic disorder in children and it is the most common of the muscular dystrophies. The dystrophin protein is associated with a glycoprotein complex at the sarcolemma. The dystrophin-glycoprotein complex connects laminin in the extracellular matrix to actin. Sarcospan is a 25kDa transmembrane component of the dystrophin-glycoprotein complex. Research from the Crosbie lab has shown that sarcospan is also a component of the utrophin glycoprotein complex, which is able to compensate for dystrophin in DMD. When overexpressed in the DMD mouse model (mdx), sarcospan increased broad sarcolemma expression of two compensatory adhesion complexes, a7b1 integrin and the utrophin-glycoprotein complex, and ameliorated pathology. Sarcospan addresses the primary defects in DMD by restoring sarcolemma stability and attachment to the extracellular matrix. Sarcospan improves eccentric muscle contraction, cardiac and respiratory function in dystrophic mice.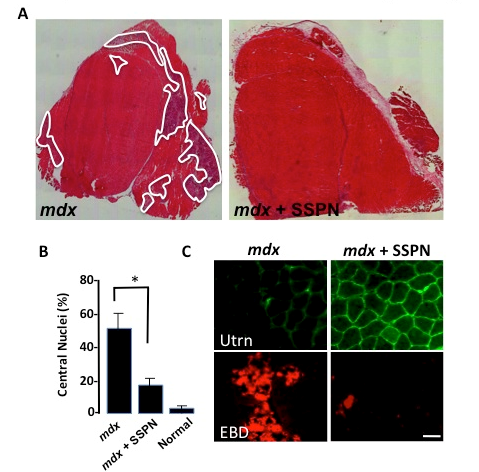
Figure: Sarcospan ameliorates muscular dystrophy in the mdx mouse model for Duchenne muscular dystrophy. (A) Transverse sections of quadriceps muscle from mdx mice revealed extensive muscle fiber necrosis (white). Overexpression of sarcospan (SSPN) eliminated muscle necrosis in dystrophic mice (mdx + SSPN). (B) Central nucleation, a marker for muscle degeneration/regeneration was dramatically decreased in mdx mice overexpressing SSPN to near normal levels. (C) Transverse skeletal muscle sections were stained with antibodies to utrophin (Utrn). Utrophin abundance is dramatically increased around the sarcolemma of individual muscle fibers in mdx mice engineered to express SSPN (mdx + SSPN). SSPN also protected the muscle from contraction-induced sarcolemma damage as illustrated with the in vivo Evans blue dye tracer assay that marks damaged (red) myofibers (EBD). Bar, 50 mm. Peter et al., 2008
The truncated dystrophin protein is present in mdx muscle. However, instead of expression at the sarcolemma, it accumulates in ER/Golgi compartments in mdx muscle where it activates the ER stress response. Sarcospan overexpression in mdx muscle decreased levels of adhesion complexes in the ER/Golgi, suggesting that sarcospan acts as a chaperone to facilitate transport to the cell surface.
The Crosbie-Watson lab is pursuing sarcospan as a target for DMD and muscle wasting using pharmaceutical and genetic approaches.