
Mutations in the X-linked gene, dystrophin, cause Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), and X-linked dilated cardiomyopathy (XLDCM). DMD and the milder form of the disease, BMD, are inherited muscle wasting disorders that affect all skeletal and cardiac muscles, while XLDCM affects only cardiac muscle. Individuals with DMD develop dilated cardiomyopathy characterized by an enlarged left ventricular chamber, thinning ventricular wall, and decreased fractional shortening and ejection fraction. Cardiac involvement in DMD has increased as a result of recent improvements in ambulation and respiratory support, which have prolonged the life of DMD patients. With the extended lifespan, development of cardiomyopathy is increasingly common in the later stages of disease. There is evidence that ACE inhibitors, steroids, and beta blockers can delay progression of heart dysfunction and development of ventricular dilation, and thus prolong the life of DMD patients. However, there is still no cure for cardiomyopathy in DMD patients. Although a number of therapeutic modalities including exon-skipping and nonsense mutation read-through strategies display efficacy and promise for the treatment of DMD, they are tailored to a select subset of patients and do not address cardiomyopathy.
The Crosbie lab is investigating a protective role of cardiomyocyte adhesion mediated by the dystrophin- and utrophin-glycoprotein complexes in response to many different physiological and pathological cardiac stressors. Furthermore, the lab is developing therapies aimed at stabilizing the sarcolemma and increasing cardiomyocyte adhesion to the extracellular matrix. It is expected that such an approach will be applicable to all DMD cases and could serve as a stand-alone therapy or in combination with any therapies that are undergoing clinical testing.
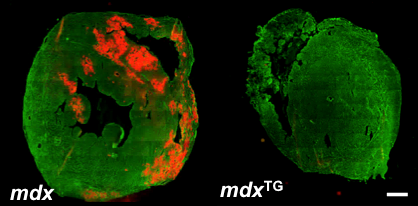
Figure: Overexpression of sarcospan attenuates membrane damage and cardiomyopathy associated with DMD. Evans blue dye (EBD) assay reveals regions of membrane instability (red) in hearts from dystrophic mice. Overexpression of sarcospan in mdx mice prevented membrane damage (mdx-Tg). Cardiomyocytes were stained with laminin antibodies (green) for visualization. Bar, 900 μm. Sarcospan also prevents cardiac fibrosis and improves left ventricular ejection fraction (LV EF%) that is characteristic of DMD (Parvatiyar et al., 2015).